Danny is a Senior Machine Learning Scientist who utilizes data-driven processes to re-imagine critical phases of the drug discovery life cycle. His primary interest is making complex biological data relatable and accessible to a broad audience to draw conclusions and base decisions. Danny brings 5+ years of in-vivo pharmacology experience from his time in the Novartis Neuroscience department. Throughout his work at Novartis and Massachusetts General Hospital, he developed skills in implementing DSP, time series, and statistical analysis pipelines for extracting translatable biomarker readouts from in-vivo electrophysiology data. He will be finishing his master’s degree in computer science from Boston University at the end of 2021.
Manual synthesis dramatically stifles innovation in drug discovery for many reasons, including (i) substantial time costs, (ii) high FTE costs, (iii) low synthesis success rates (20-34%), and (iv) a bias towards known reactions.
At DeepCure, we are fixing these problems to unlock the vast chemical space that AI drug design tools want to explore, but don’t, because manually synthesizing such compounds would not be practicable.
Automated Robotic Custom Synthesis
Manual synthesis dramatically stifles innovation in drug discovery for many reasons, including (i) substantial time costs, (ii) high FTE costs, (iii) low synthesis success rates (20-34%), and (iv) a bias towards known reactions.
At DeepCure, we are fixing these problems to unlock the chemical space that AI drug design tools want to explore but can’t because it is not practicably available to most chemists.
Synthetic
Steps
Reaction
Types
Reaction
Development
Industry Standard
1
4-10
manual
91
100+2
fully
automated
made possible with automated analytical evaluation, purification, evaporation, etc.
made practicable with automated reaction development
made feasible with miniaturization & quick turnaround (2-10 days)
Notes: 1 4+ by Q2 2024. 2 Planned to reach by end of 2024.
MolGen™
Our molecular generation tool, MolGen™, designs novel, diverse compounds. Using state-of-the-art deep reinforcement learning (RL), MolGen™ constructs synthesizable compounds with features that capture the important molecular interactions for binding and selectivity, as well as deliver the desired ADME-tox profile of the target candidate profile (TCP).
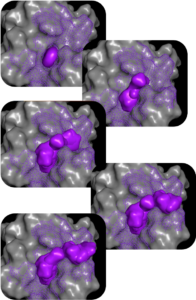
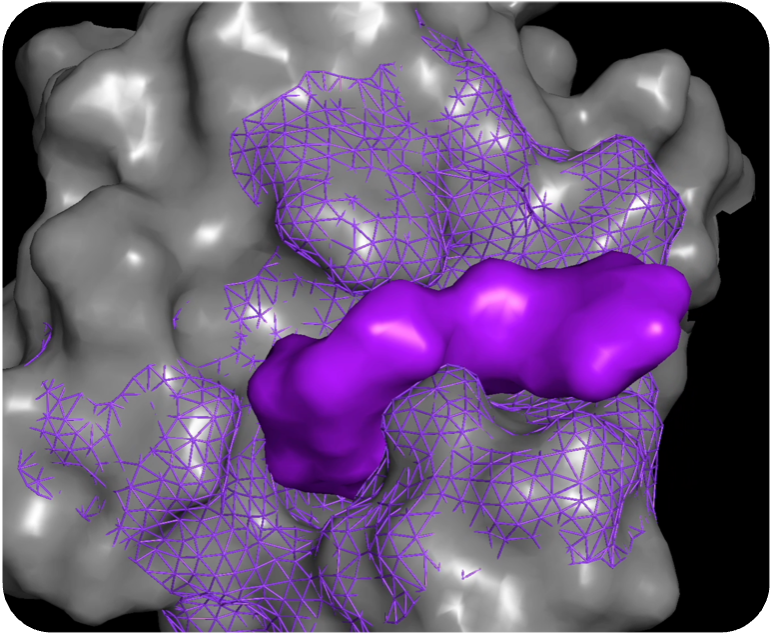
Output of PocketExpander™
MolGen™ – building & iterating compounds
Novel, potent, & selective compound
Hypothesis Generation
Unlike other AI drug discovery companies, DeepCure does not use AI to simply match a library of compounds to a known pocket. Instead, we use our patent-pending AI methods to create causal, data-driven, human-interpretable hypotheses for binding to a given protein target. This enables us to go beyond known binding sites and ligands.
Structure-Based
Our hypothesis generation starts with a rigorous analysis of available structural information. Beyond the standard steps involved in structure preparation, our proprietary protocols also include methods for repairing structures (e.g. building missing loops) and generating more robust structures leveraging molecular dynamics (MD).
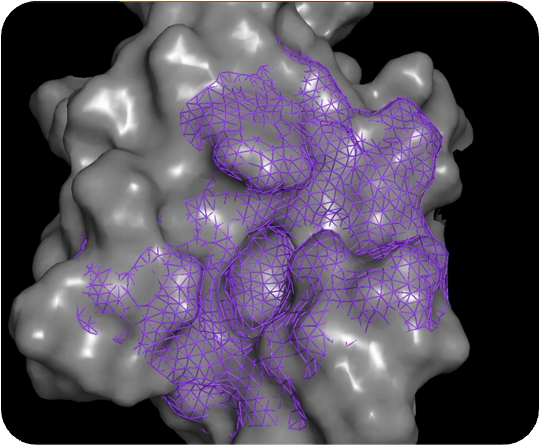
PocketExpander™
For most therapeutic targets, there is no data, limited data, or biased data. PocketExpander™ allows us to generate novel hypotheses by leveraging AI/ML and computational chemistry methods to map the protein surface and identify novel binding modes (shown as colored dots). The outputs serve as the blueprints for our molecular generation tool, i.e. MolGen™.
Causal Analysis
ML methods for drug discovery typically focus on correlations, which lead to biases for the types of compounds that have previously failed in discovery. In contrast, DeepCure uses a causal ML approach to find binding interactions without the biases for fruitless binding modes to design truly novel compounds.
Medicinal chemists engage in a conversation with explainable models
DeepCure’s platform is designed to be human-interpretable. By seeing how molecules are predicted to interact with the protein, scientists can make rational design changes to the molecule and explore interesting molecular interactions – ensuring we don’t blindly follow the ML algorithm or chemists’ intuition.