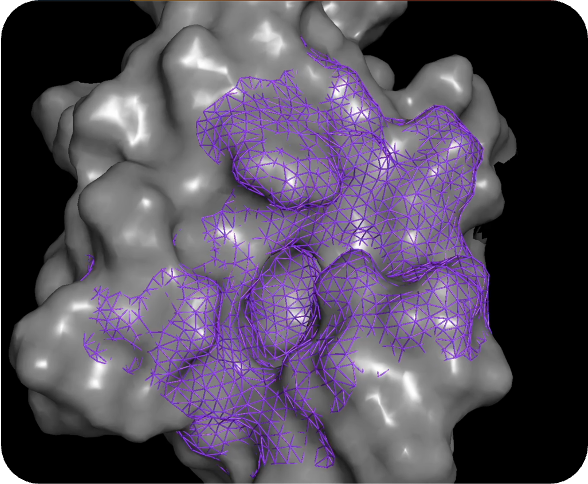
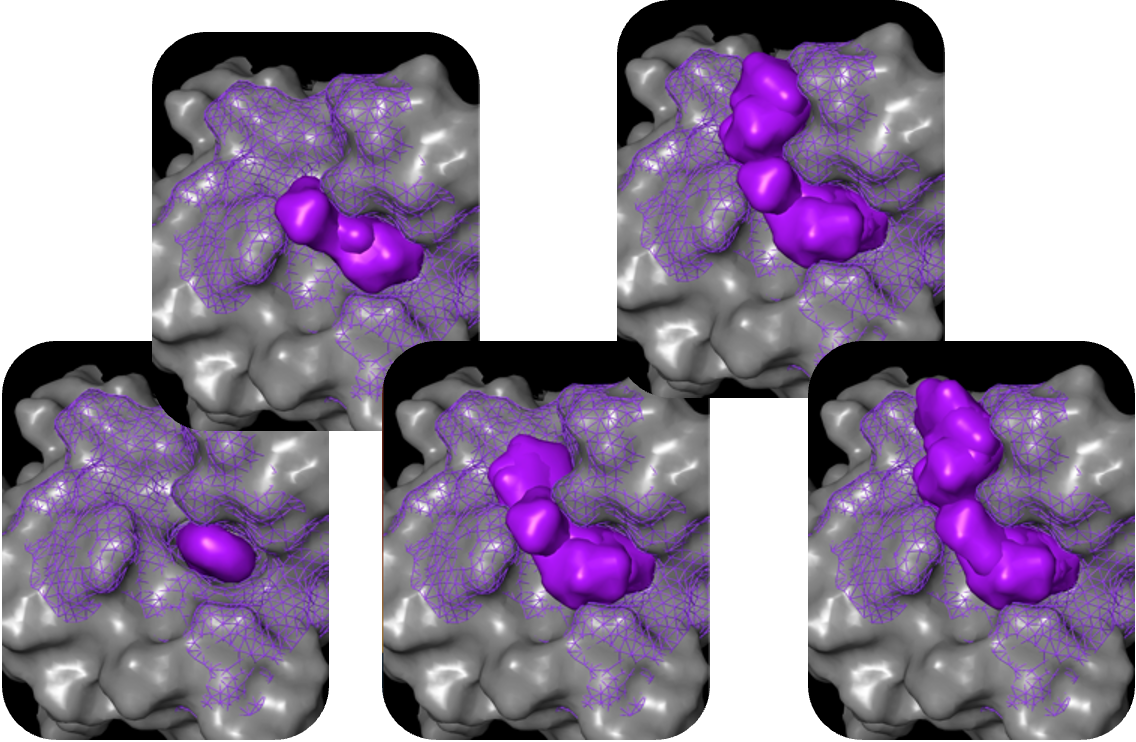
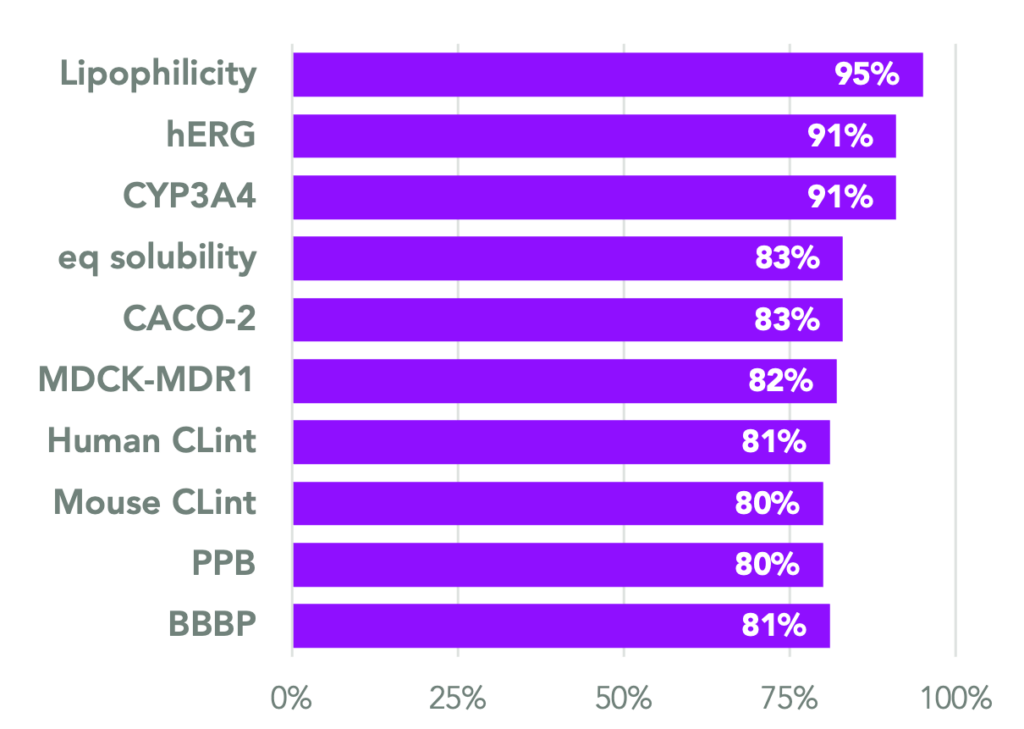
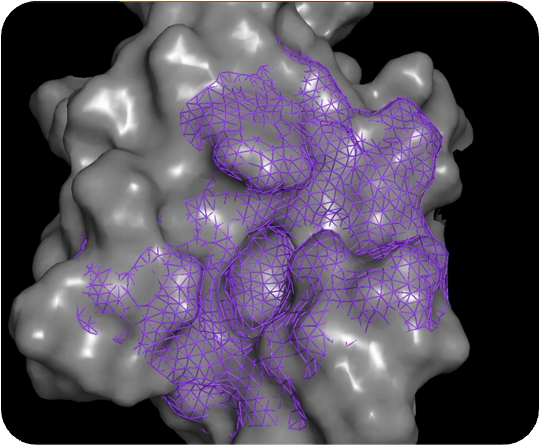
Our patent-pending molecular generation tool, MolGen™, is used to design novel, diverse compounds that are readily synthesizable and fit the desired TCP for a given drug program.
The molecular generation process leverages state of the art deep reinforcement learning (RL) to construct synthesizable molecules that efficiently capture all causal interactions for binding and selectivity while maintaining the desired ADME-tox profile.
MolGen™ utilizes hundreds of workers, choreographed in a unified pipeline (leveraging distributed training, hyperparameter optimization, GPU instances, and more), to improve the output quality of molecules as quickly as possible.